トランスジェニックマウスの導入遺伝子のgenotyping
トランスジェニックマウスの系統維持を行う上で重要な遺伝子型判定の方法について解説する(Noguchi et al., 2004)。 ちなみに本法は、元々は「近傍プライマーPCRによるzygosity check法」のために開発したのだが、 本法では挿入遺伝子近傍のゲノム配列を利用するので、その配列情報をもとに 近年整備されたゲノムデータベースを活用すると、挿入遺伝子近傍配列が染色体上のどこにあるかを特定できることがあり、 その意味で挿入遺伝子のマッピングにも利用できる技術である。 さらに挿入遺伝子の挿入パターンも検出できることもあり(Suzuki et al., 2006)、遺伝子導入動物の解析を行う上で便利なシステムである。
この方法についてのご質問・ご意見等についてはこちらまでご連絡いただきたい。できるだけ対応したい。
導入遺伝子のZygosity(ホモ・ヘミ)の判定法
遺伝子導入動物の導入遺伝子のホモヘミ判定には、いろいろなやり方がある。例えばトランスジェニックマウスを作る上でのバイブル的存在である本「Manipulating the mouse embryo (3rd ed.)」には以下のような判定法があげられている。
- 遺伝子量を測る
- サザンブロット解析
- Realtime-PCRによる定量
- 遺伝子産物の量を測る(酵素活性など)
- FISHによる核型の判別
- テスト交配
- 近傍プライマーによるPCR
遺伝子型の判定は、遺伝子導入動物の維持や使用に当たって必ず、かつ頻繁になされるので 可能ならば、ルーチンでやれるように簡便で安全で確実なモノが好ましい。 全個体について毎回サザンブロットやFISHを行うのはかなり手間がかかるし、 Realtime-PCRで「確実に1倍量と2倍量を見分ける」にはサンプル調整を厳密に行う必要がある。 一方、定量的にではなく定性的に判定できれば、DNA濃度調整などがラフに行えるので汎用性が高い。 その意味で、定性的な判断が可能な「近傍プライマーによるPCR」が実用的である(詳細は後述)。 遺伝子導入動物を使う場合、導入遺伝子のあるなしを必ずPCRか何かで確認する必要があるわけで、 その際ついでに「ホモ・ヘミ」までわかってしまえば非常に便利であると言える。 ただし、一般に導入遺伝子は、ゲノムのどの位置に挿入されるかを予見することは難しいため、 何らかの方法で導入遺伝子近傍の配列を求め、しかる後に近傍プライマーを設計することが 必要となる。この手間があるので「簡便ではないではないか」という批判もあるが (実際はそれほどの手間ではないのだが、一度もやったことがない人がいきなり始めるのは大変かもしれない)、 一度確定してしまえば例えば毎回サザンブロットをやるよりは遙かに楽になるので一考の価値はあると思う。
近傍プライマーを用いたPCRによる遺伝子型判定
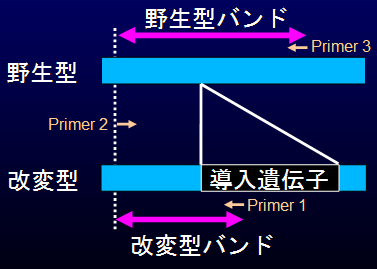
この方法は、右図で示すように、導入遺伝子のゲノム挿入位置を跨ぐようなプライマーを3つ同時に使用し、
PCR産物の長さと組み合わせで導入遺伝子についてホモ型(+/+)・ヘミ型(+/-)・野生型(-/-)を判定するというものである。
他の多くの方法が何らかの定量を行わねばならないので、テンプレート量を揃えたり、
内部標準を用意したりなどの手間がかかるが、
この方法はバンドのあるなしの組み合わせで判定できるので、
それほど量にこだわらなくてよい利点がある。
ただ、この方法の難点は、PCRの宿命でもあるのだが、
「ゲノムとの接合部(=PCRで増幅される部分)しかモニターしていないので、
注目している場所以外の場所に変異が入ってしまったら判定と合わなくなる」という危険性があり、
実際に動物を使う際には導入遺伝子の発現(mRNAやタンパクの発現、または表現型の発現など)を
確認して、きちんと導入遺伝子が存在し、機能していることを確認する必要がある。
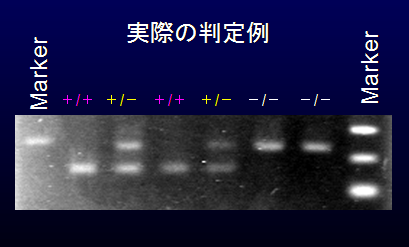
実際の判定例を右に示す。
この例では、長いバンドが野生型由来(-)、短いバンドが改変型由来(+)として
検出されるようにプライマーが設計されており、
実際にバンドの長さと本数の組み合わせでホモ・ヘミが明確に判定できていることがわかる。
ここで重要なのは「各個体で必ずPCRのバンドが1本以上現れる」という点である。
単純に「バンドが全くでないのが陰性でバンドがでれば陽性」という基準で判別すると、
「PCR自体に問題があって全く産物ができなかった」という可能性を排除できない。
極端な話、「テンプレートを入れ忘れた」だけで「陰性判定」になってしまう。
そこで、本法では必ず野生型由来か、改変型由来か、もしくは双方のバンドが増幅されるように設定してある。
もし「全くバンドがみられない」場合はPCR自体が不良と言うことを意味し、その場合は再試験をすることになる。
Flanking primerの設計方法
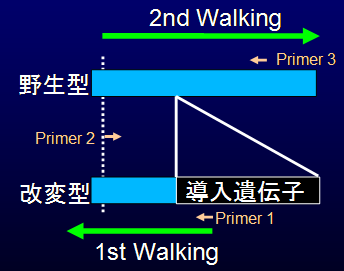
Genomic Walking(後述)により導入遺伝子の近傍配列を求め、挿入位置を跨ぐようなプライマーを設定する。
手順1) 1st Walking (Primer 1を起点)
導入個体ゲノムを用いて導入遺伝子を基に5'-近傍配列を決定し、Primer 2を設計する。
手順2) 2nd Walking (Primer 2を起点)
親系統ゲノムを用いて5'-近傍配列を基に3’-近傍配列を決定する。実際には、データベース情報が活用できる場合がある。
手順3) Flanking primerの設定
3'-近傍配列を基にPrimer 3を設計する。
Genomic Walkingによる近傍配列の決定
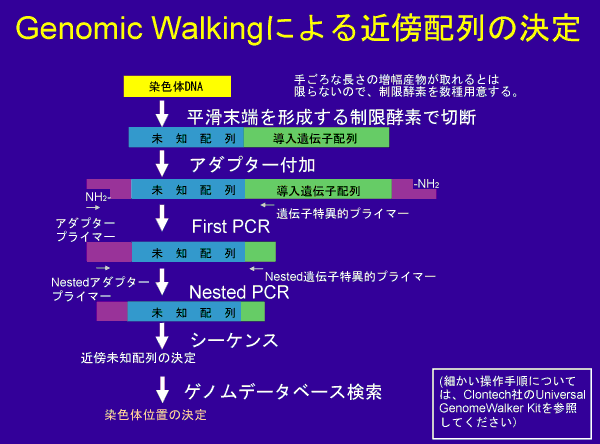
ここで示す方法は、Clontech社のUniversal GenomeWalker Kitによる方法である。 PCRをベースにしたシステムを用いて未知の部分を増幅しようとする場合、 「いかにして未知の部分にプライマーを設定するか?」が問題となるが、 そのキットでは、制限酵素処理によってゲノムを断片化し、 その断端にアダプター配列を結合することによって アダプタプライマーがアニーリングできる場所を導入するという戦略を使う。 ただし、PCRでうまく増幅できるような場所にアダプターが導入される確率は それほど高くはないため、複数の制限酵素を用いて、複数のアダプター結合ライブラリを作る必要がある。 ちなみにそのキットでは、4種類の制限酵素により4つのアダプター結合ライブラリを作るが、 我々の研究室では念のため酵素を6種類にしている。 導入する遺伝子によっては、その内部に使用しようとする制限酵素部位がある場合もあるので、 その場合は導入遺伝子がタンデム挿入されていた時(これが一般的)に必ず、しかも多数コピー由来の断片が 制限酵素により生成されてしまうので避けた方がよい(その場合、厳密には偽陽性というわけではないが その断片に由来するPCR産物にはゲノム情報は含まれず、近傍配列決定にはnon-informativeであるため、 できればそうした増幅産物の生成は避けたい)。 そういう意味で実際に用いる酵素の選択も非常に重要である。 基本的にはアダプターの結合部位が平滑末端なので平滑末端を生じる制限酵素を用いるが、 平滑末端生成酵素でなくても、平滑化処理をすればいいので、 実際は任意の制限酵素が使用可能である(この点については実際に実施可能であること確認した)。 また、厳密度を上げられるようにプライマーの長さをキットに同封されているものよりも若干長めにして、 なるべくきつい条件でPCRを行えるようにして改変して用いている。
実施に当たってのポイントとしては、極力偽陽性を避けるための方策をとるべきである。例えば、
- いわゆるホットスタート酵素(化学修飾型が望ましい)を用いたHot Start PCRによりPCR精度を上げる。
- いわゆるShuttle PCR法を用いて、アニーリングステップを高い温度にしたPCRを行う。
- 長めのプライマーにより(27~29-mer)、アニーリングステップを高い温度に設定できるようにする。
- 必ずnested PCRを行って2組4本のプライマーにより認識・増幅されるバンドを検出する。
この方法で、かならず近傍ゲノム配列が求まるか、というとそれはまさに「確率との勝負」である。 挿入遺伝子はどこに挿入されるか予見できず、PCRで増幅可能な距離に制限酵素の切断部位があるかどうかはやってみなければわからないし、 得られた配列が非常に特異性が低く、適切なプライマーを設定できない場合もあり得る。 外来遺伝子が挿入される際には、その周囲の配列にも変化が生じることもあり、 ゲノムデータベースで全く見つからない配列が挿入されている例もある。 しかもゲノムデータベースのカバー率も現時点(2005年)では100%ではないという事情もある。 しかし、近傍ゲノム配列が確定できた場合の利点は非常に大きく(今までの成績では成功率は7割程度)、 この方法を試してみる価値は十分あると思う。
実際例
実際の適用例について以下に記載する。
pCAGGS vectorを基にした導入遺伝子コンストラクト
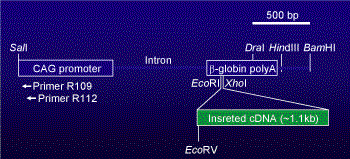
当研究室では、主にpCAGGSベクター(Niwa et al., 1991)を基本として遺伝子導入用コンストラクトを構築している。 pCAGGSベクターは、いわゆるCAGプロモーター、すなわち、Cytomegalovirus enhancerと Chicken β-actin promoterをつなげた構造と、兎のβ-グロビン遺伝子のpolyA signalサイトによって、 導入したい遺伝子をほぼ全身性に過剰発現させることができる。
Genomic Walkingを行う場合は、基本的にはホストDNAとはホモロジーが低いであろう部分、 すなわち非哺乳動物性遺伝子部分にプライマーを設定すると特異性が高いので、 CMVエンハンサー部位にWalking primerを設定している(R109とR112として示した場所)。
Genomic walkingによる近傍配列の決定
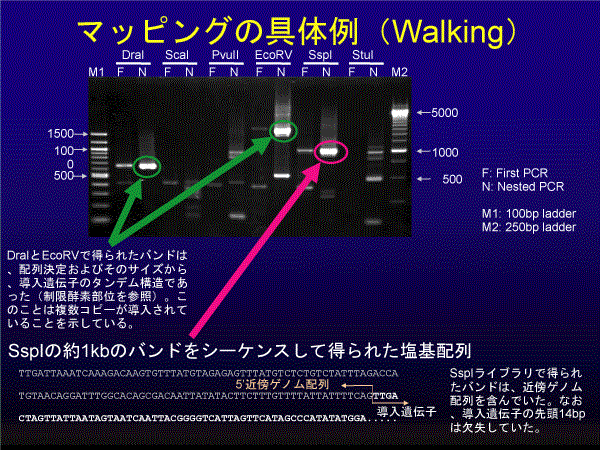
4C30系マウスで実際にGenomic walkingを行った例を示す。理想的なのは、FirstPCRと Nested PCRでともにバンドが検出されているものをシーケンスすることであるが、常にそのようなバンドが得られるとは限らないので、 試行錯誤がある程度必要かもしれない。濃いバンドの場合は、導入遺伝子がタンデムに連結している場合が多い(今回のDraIや EcoRVで見られたバンド)。この例ではSspIライブラリで近傍配列が含まれたバンドが得られた。
Ensembl databaseによる染色体位置の検索
得られた近傍配列を元にEnsemblデータベースのBlast検索で染色体位置の検索を行う。
この画面は現在はもっと新しいものになっているが、今回の例で使った時点での画面の例を挙げておく。
なお、データベースは日々更新されているので(2006年4月の時点でversion 38)、検索を行った際はバージョン番号も記録しておくべきであろう。
染色体位置の判定例
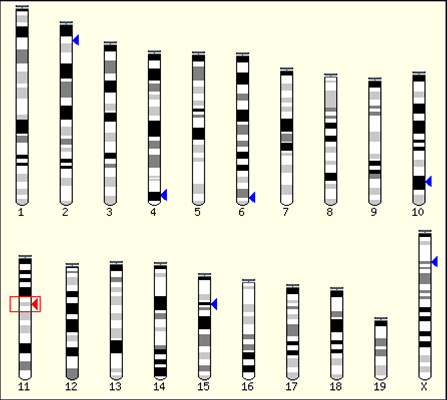
今回の例でEnsemblデータベースで得られた導入遺伝子の染色体上の挿入部位を示す。11番染色体に導入遺伝子の近傍配列が存在する、すなわち導入遺伝子は 11番染色体上に挿入されたことがわかる。さらに詳しい位置関係もデータベースで知ることができる。 データベース検索の初期設定では可能性の低い位置も候補として表示されるので、実際にはスコアの基準値を設定して可能性の高い部分に絞り込むとよい。
データベースでヒットした部位の周辺の配列をデータベースから得ることができるので、その配列を元に近傍プライマーを設定すれば、上記であげたようなホモ・ヘミ判定用プライマーが設定できる。ただし導入遺伝子の挿入パターンには複数考えられ、いろいろと考慮しなくてはならない点が多い。 これらの議論についてはNoguchi et al., 2004で考察しているので参照していただきたい。
参考文献
- Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108(2):193-199. [PMID: 1660837]
- Noguchi A, Takekawa N, Einarsdottir T, Koura M, Noguchi Y, Takano K, Yamamoto Y, Matsuda J, Suzuki O. 2004. Chromosomal mapping and zygosity check of transgenes based on flanking genome sequences determined by genomic walking. Exp Anim 53(2):103-111.
- Suzuki O, Hata T, Takekawa N, Koura M, Takano K, Yamamoto Y, Noguchi Y, Uchio-Yamada K, Matsuda J. 2006. Transgene insertion pattern analysis using genomic walking in a transgenic mouse line. Exp. Anim. 55(1):65-69.